
Folding delle proteine
FOLDING (RIPIEGAMENTO) DELLE PROTEINE
DOGMA CENTRALE: La sequenza specifica la struttura.
La struttura finale è quella con più bassa energia interna, in assoluto o cineticamente accessibile.
1) Il paradosso del folding (Levinthal):
Una proteina con 100 residui e 3 possibili conformazioni per residuo (N° molto limitato) ha 3100 possibili conformazioni.
Supponiamo che transitare da una conformazione ad un'altra, per poterle saggiare ciascuna, una dopo l'altra, siano necessari 10-13 sec. (tempo medio per rotazioni), la scansione di tutte le possibili strutture richiederebbe 1027 anni (ben oltre la vita dell’universo). E' evidente che le proteine non si ripiegano saggiando casualmente tutto lo spazio conformazionale disponibile.
Il tempo di ripiegamento si riduce notevolmente se ogni qual volta che un residuo trova un elemento della conformazione corretta, lo conserva nel prossimo tentativo. Una caratteristica essenziale del processo di ripiegamento è quindi la conservazione di elementi parzialmente corretti.
2) Denaturazione/rinaturazione
La struttura funzionale di una proteina, la sua 'struttura nativa', è quella energeticamente più stabile. Una proteina si denatura quando sono interrotte le interazioni multiple non-covalenti che stabilizzano la struttura nativa. Il processo è cooperativo, poiché la rottura di un legame non-covalente (es. un legame-H) facilita la successiva rottura di un secondo legame, e così via. Essendo cooperativo da luogo curve di denaturazione di tipo sigmoide (Fig. 1).
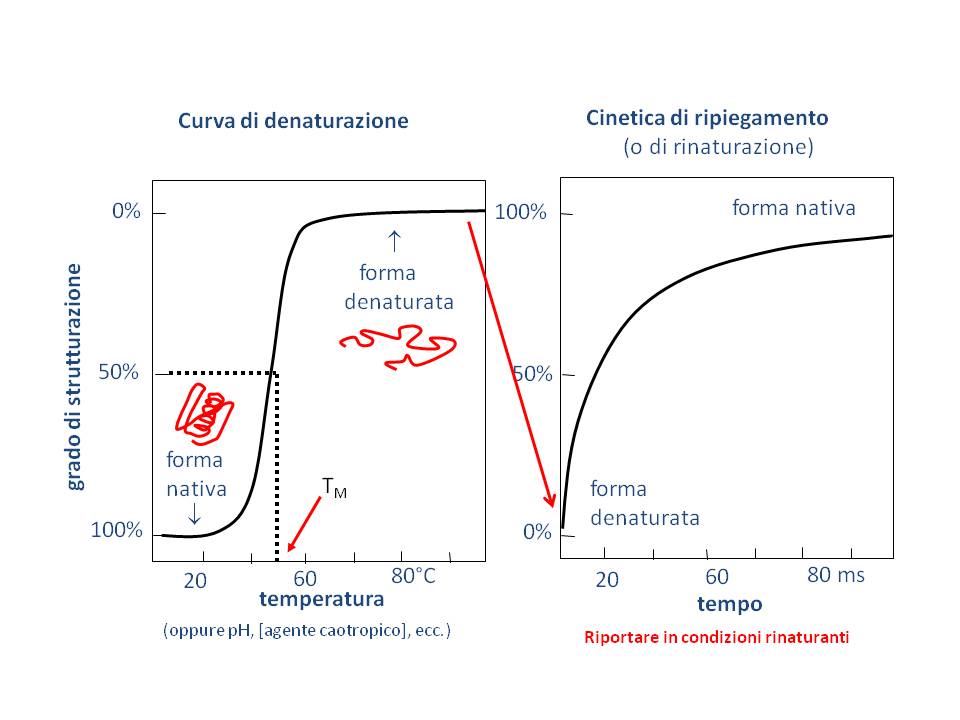
Fig. 1 Curve di denaturazione e di cinetica di rinaturazione
La denaturazione può essere causata da un aumento di temperatura, da variazioni nel pH o della forza ionica, o anche da agenti caotropici se presenti ad elevate concentrazioni (vedi Fig. 2)(vedi anche la pagina Macromolecole Biologiche). La temperatura di fusione (Melting temperature, TM, Fig.1), è quella alla quale la metà dei residui sono destrutturati. In linea di principio, se i fattori che causano la denaturazione sono rimossi (es. si raffredda la soluzione o si riduce la concentrazione degli agenti caotropici), e non ci sono altri fattori di disturbo, la proteina si rinatura seguendo la curva a ritroso. Se il processo di denaturazione/rinaturazione può essere seguito nel tempo, si ottengono curve per la le cinetiche di folding (vedi Fig. 1 a sinistra).
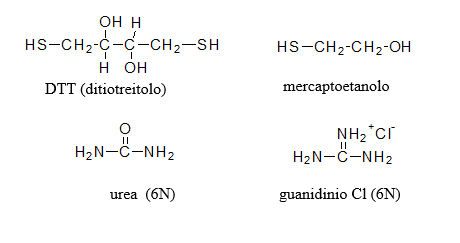
Fig. 2 Agenti che riducono i ponti disolfuro (DTT,
mercaptoetanolo) e agenti caotropici (urea, guanidinio cloruro)
La denaturazione di una proteina causa forti variazioni negli spettri CD e di fluorescenza (Fig. 3). Nel primo caso ciò è dovuto alla transizione da conformazioni regolari a random coil; nel secondo caso al passaggio dei residui di triptofano da un ambiente interno più organico e protetto ad un ambiente acquoso. Queste due tecniche sono quindi molto utilizzate per seguire processi di denaturazione / rinaturazione in tempo reale.
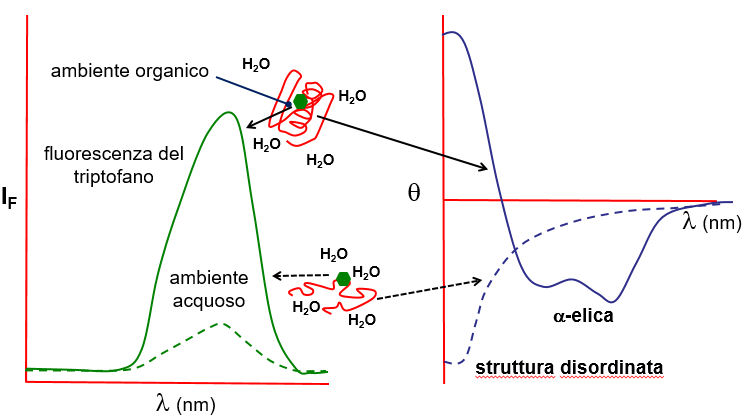
Fig. 3 effetto della denaturazione sugli spettri di fluorescenza e CD delle proteine
COSA SPINGE IL RIPIEGAMENTO ?
1) Per piccole proteine, Anfinsen ha proposto che il ripiegamento avviene spontaneamente e direttamente alla struttura più stabile, che è quella nativa (Fig 4, centro). I residui idrofobici, in particolare, cercano di segregarsi all'interno della proteina per evitare l'ambiente acquoso. Per subunità proteiche con dimensioni maggiori, un altro modello per il folding propone che questa necessità promuove inizialmente il “collasso idrofobico” della catena estesa e non strutturata, con formazione del cosiddetto “globulo fuso” (tempo millisecondi). Questa è una struttura compatta anche se ancora disordinata e molto dinamica, nella quale i residui idrofobici sono rivolti verso l'interno e protetti dall'ambiente acquoso.
2) Alcuni segmenti della catena iniziano ad adottare conformazioni ordinate per poter formare legami-H. Queste “unita ripieganti autonome” sono transitorie, ma fungono da centri di 'nucleazione' per la formazione di elementi di struttura ordinata più stabili. All'interno del globulo fuso la necessità di formare legami-H fra tutti i gruppi amminici e carbossilici favorisce la formazione di struttura. Si formano intermedi strutturali metastabili, che eventualmente danno luogo alla struttura terziaria finale (tempo da secondi a minuti).
3) Secondo altri modelli, “unita ripieganti autonome” possono formarsi a prescindere dal collasso idrofobico, e portare alla formazione della struttura finale stabile (vedi Nucleation growth e Framework models nella Fig. 4). Un altro modello suggerisce che siccome le molecole di proteina disordinate hanno strutture di partenza anche molto diverse fra di loro, potrebbero ciascuna seguire un diverso percorso, che si conforma più o meno ad uno o l'altro dei modelli citati in precedenza (Jigsaw model).
4) La struttura finale non è determinata dalla sequenza esatta di residui, quanto al pattern di differenti tipi di residui (polari idrofobici, carichi ecc.). Quindi, sequenze anche molto diverse possono dare luogo a strutture terziarie simili, sempre che sia mantenuto un pattern simile di residui idrofobici ed idrofilici (un esempio sono le emoglobine di diversi animali, che hanno dimensioni e strutture simili ma sequenze assai differenti). Questo è intuitivamente ragionevole, in quanto esiste un numero elevatissimo di proteine con diverse sequenze, ma un numero limitato di diversi tipi di struttura (si calcola fra 1000 - 5000 diverse topologie).
5) Per proteine molto grandi, si ripiegano per prima domini strutturali, oppure le subunità di struttura terziaria, e per ultimo avviene l'associazione dei domini nella struttura finale o l'aggregazione delle subunità in oligomeri con struttura quaternaria.
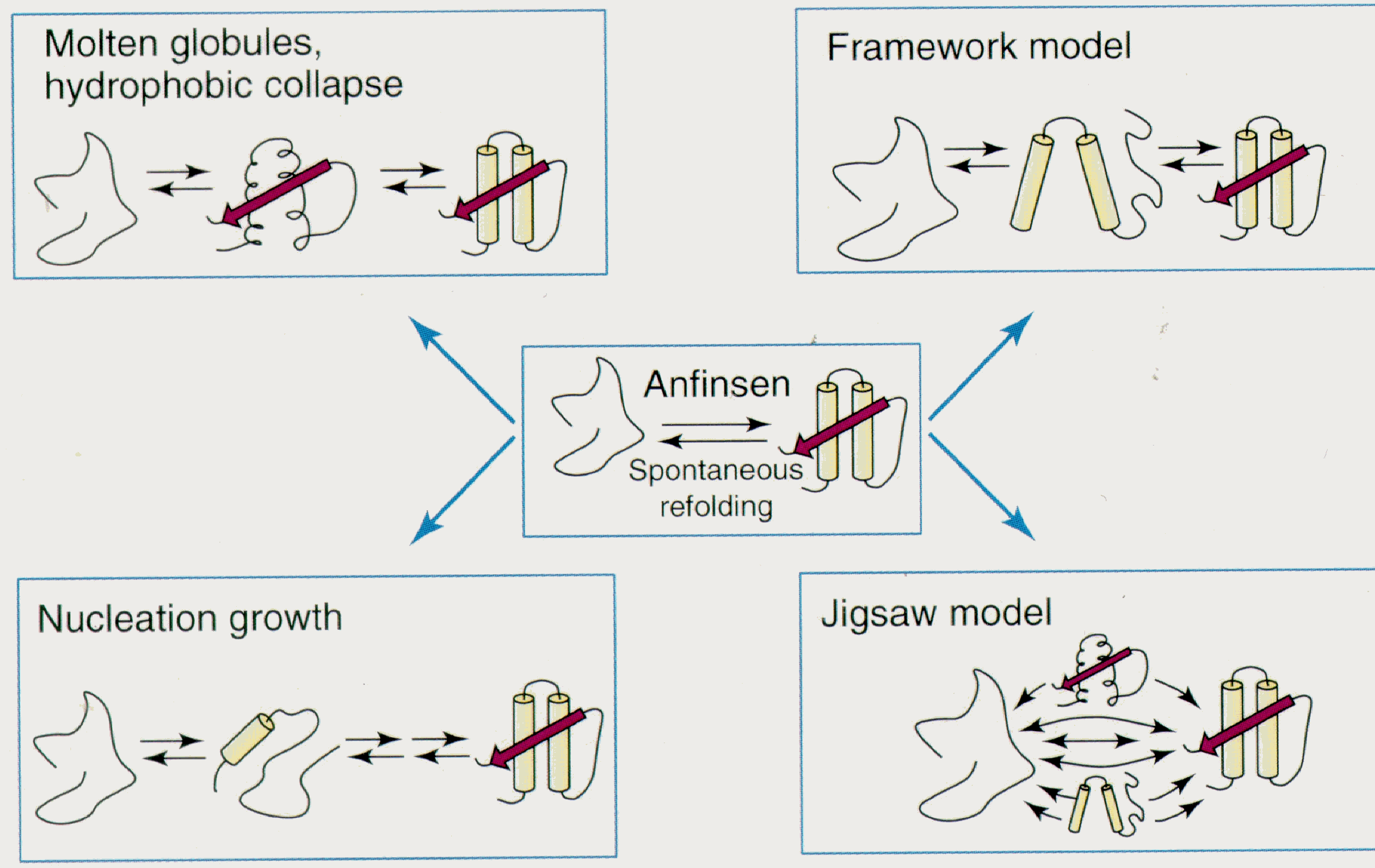
Fig. 4 Modelli per il ripiegamento di proteine
6) In conclusione, le catene proteiche non ripiegate hanno strutture disordinate in molte diverse maniere. In altre parole, coprono uno spazio conformazionale disordinato molto ampio, e hanno un elevato contenuto energetico. Questo si può schematizzare come il plateau ad alta energia nella Fig. 5. Quando iniziano a ripiegarsi, entrano in un cosidetto "folding funnel" una versione del 'panorama energetico molecolare' che presuppone che lo stato nativo della proteina corrisponde ad un minimo nell'energia libera. La parete di questo imbuto (cioè la traiettoria energetica seguita dalla catena durante il ripiegamento) può essere assai "accidentata". Sono presenti molti minimi energetici locali, che corrispondono a strutture non ‘native’ nei quali le catene parzialmente ripiegate possono 'intrappolarsi'. Da questi minimi escono sfruttando l’energia termica, oppure con l’aiuto di altere proteine (vedi sotto). La teoria del ‘folding funnel’ presume che lo stato nativo sia un minimo molto profondo, con pareti ripide, corrispondente ad una specifica e ben definita struttura terziaria, dal quale la catene non può uscire. Molte proteine, in assenza di aiuto da parte di altre proteine, predisposte per assistere il folding, entrerebbero in profondi minimi che corrispondono a strutture non-native, e queste 'misfolded proteins', oltre non essere funzionali, potrebbero essere dannose per l'organismo.
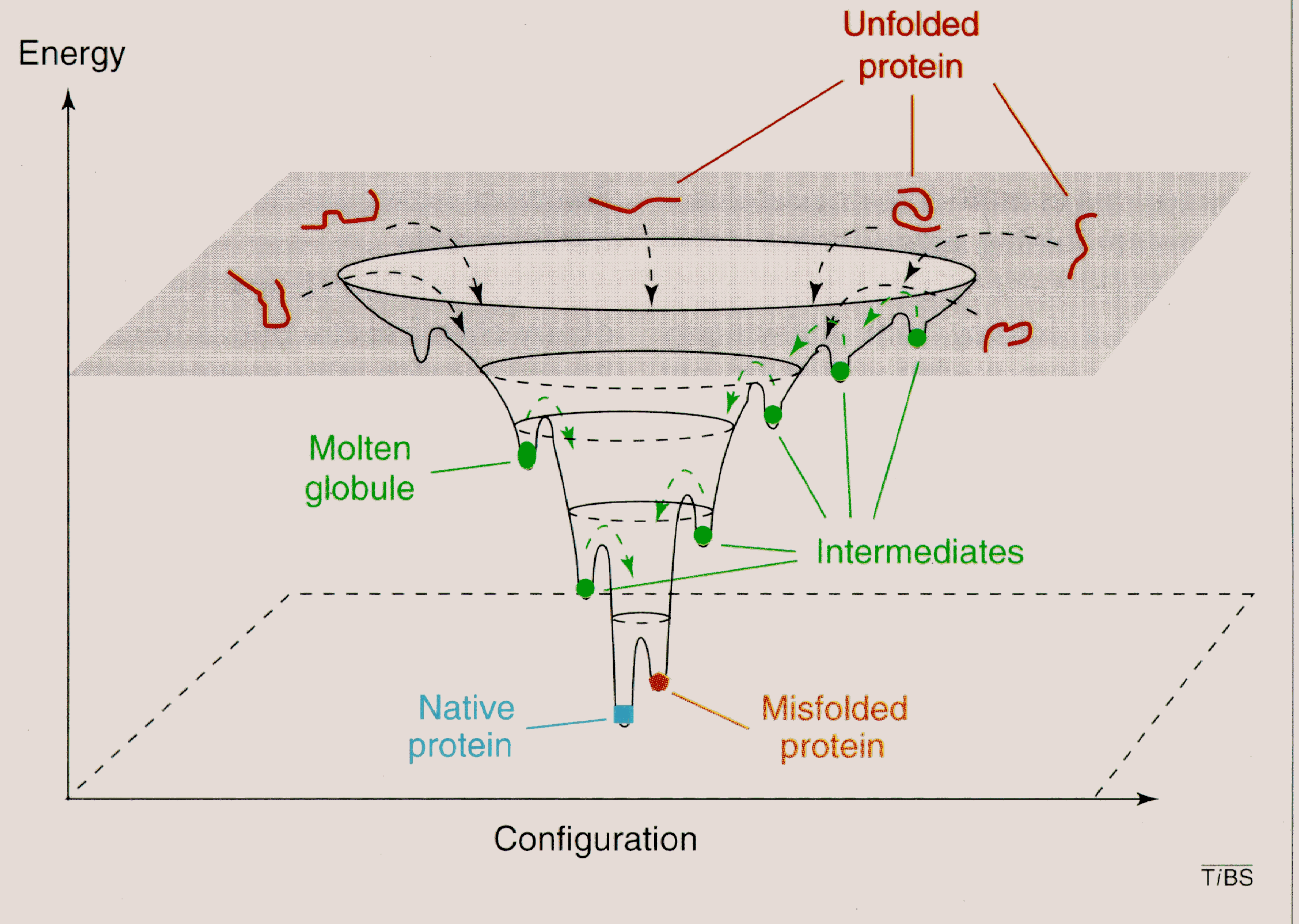
Fig. 5 Panorama energetico per il ripiegamento di una proteina; la teoria del "Folding funnel"
PROTEINE CHE AIUTANO IL PROCESSO DI RIPIEGAMENTO
* Il ripiegamento di proteine può essere notevolmente rallentato da residui di Prolina in configurazione incorretta (cis/trans) o di Cisteina coinvolti in legami S-S non corretti, poiché la conversione nella forma corretta è un processo lento. nella cellula, sono presenti proteine che hanno come funzione quella di promuovere il corretto ripiegamento, per esempio velocizzando la conversione di PRO e Cys alle forme strutturali corrette, oppure impedendo che durante il processo di ripiegamento, le catene non ancora ripiegate interagiscano con altre catene per formare aggregati amorfi e non funzionali.
Alcune ISOMERASI catalizzano l' interconversione dei legami cis/trans nella prolina, o rottura/riformazione di ponti disolfuro nella cistina, e quindi velocizzano il processo di folding:
- Le Cis/trans o Peptidilprolil isomerasi sono proteine ubiquitarie e molto abbondanti, con svariati ruoli oltre a quello di interconvertire legami peptidici cis/trans (es la ciclofillina è coinvolta nell'immunospressione)
- Le proteindisolfuro isomerasi catalizzano l’interconversione di ponti disolfuro permettendo la formazione della struttura corretta, che essendo quella più stabile poi permane. Contengono residui di cisteina molto reattivi che spiazzano uno dei due partner nei ponti disolfuro delle proteine bersaglio. Formano transitoriamente ponti intermolecolari e poi rilasciano i residui di cisteina delle catene substrato, permettendole di riformare nuovi ponti intramolecolari nella catena correttamente ripiegata.
- Le Chaperon molecolari impediscono interazioni dannose con altre catene. Il ripiegamento è rallentato o anche completamente impedito se la catena proteica non ancora strutturata interagisce con altre catene proteiche. Infatti, le catene proteiche in fase di ripiegamento sono piuttosto appiccicose, essendo i residui idrofobici ancora parzialmente esposti.
Quando una catena proteica viene estrusa dai ribosomi (sono sintetizzate dal N-terminale), in maniera lineare e vettoriale, un corretto ripiegamento non è possibile fino a che non sia estruso almeno un intero dominio strutturale. Prima che ciò avvenga, la catena estrusa è esposta e molto suscettibile alle interazioni non desiderate. La concentrazione di proteine all'interno della cellula è molto elevata (dell'ordine del millimolare), ed in proporzione, quindi anche quella di catene non-strutturate (si stima dell’ordine di 35 micromolare in E. coli).
Il corretto ripiegamento può quindi richiedere la presenza di proteine che aiutano la catena polipeptidica crescente ad evitare interazioni dannose. Esse sono note come Chaperon molecolari. Non fanno parte della struttura finale delle proteine che aiutano a ripiegare, e non aumentano la velocità di ripiegamento; semplicemente evitano che avvengano interazioni illecite che portino a forme di ripiegamento incorrette.
GroEL e GroES.
E’ stato definito il meccanismo d’azione della chaperonina batterica GroEl/GroEs, con la recente determinazione della sua struttura cristallografica ad alta risoluzione.
1) GroEL è composta da 14 subunità poste in due anelli di 7 subunità ciascuna, denotati anelli cis e trans. E’ un complesso proteico di forma cilindrica, con dimensioni 150Å x 140 Å, e poro centrale di < 50 Å. (Fig. 6). Ciascuna subunità contiene un sito di legame per catene polipeptidiche ed un sito di legame ed idrolisi di ATP. Il sito di legame per le catene polipeptidiche consiste in una superficie idrofobica alla quale le catene non ripiegate, che espongono residui idrofobici, si possono appiccicare in maniera non-specifica .
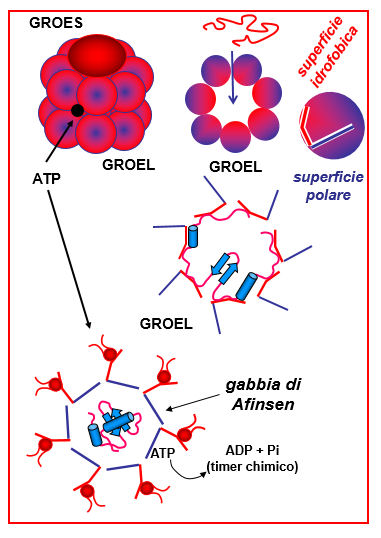 .
. 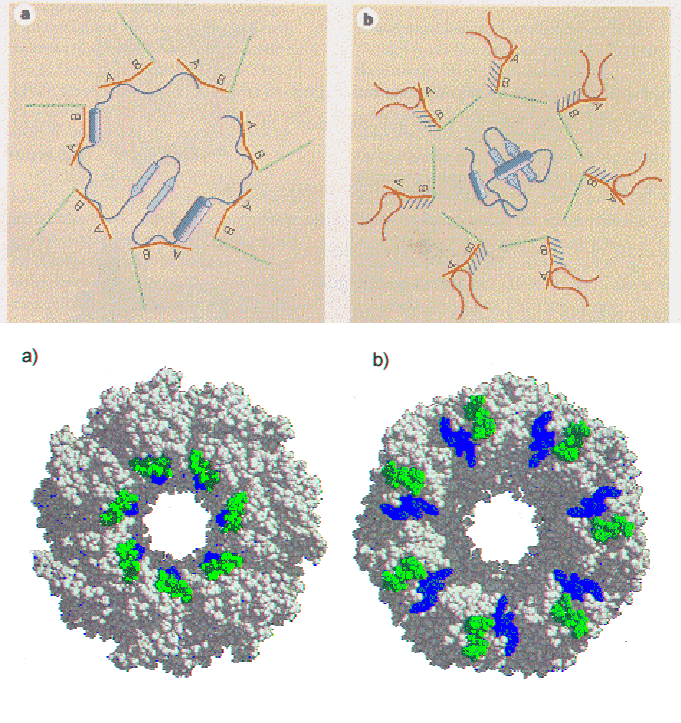
Fig. 6 Struttura schematica e meccanismo d'azione della chaperonina GroEL/GroES. Nel pannello in alto a destra visualizzazione schematica della variazione conformazionale che porta alla formazione della 'gabbia di Anfinsen'. In basso a destra visualizzazione molecolare dall'alto della moleocla con indicata la superficie polare che si sposta via dal poro quando si forma la gabbia
2) Una catena parzialmente ripiegata entra nel poro da uno lato di GroEL, e su quel lato poi si lega una proteina a forma di duomo, GroES, a mò di coperchio. Questo isola la catena polipeptidica nel lumen del poro.
- Superfici idrofobiche nelle subunità che formano gli anelli di GroEL foderano le pareti del poro, che ha un diametro di ca. 25 Å. Queste superfici attirano i residui idrofobici nella catena polipeptidica parzialmente strutturata.
- Contemporaneamente, si legano 7 molecole di ATP, una per ciascuna subunità dell'anello, e ciò causa una transizione conformazionale in GroEL che fa allargare il poro a 35 Å. Questa espansione causa la completa destrutturazione della catena appiccicata alla sua superficie interna.
- La transizione conformazionale sposta anche le superfici idrofobiche che foderavano l’interno del poro, che adesso diventa idrofilico. La catena polipetidica substrato viene quindi rilasciata nella cavità interna (lumen) del poro, la cosiddetta “Gabbia di Afinsen”, dove gli è permesso di ripiegare un ambiente completamente protetto.
4) La transizione conformazionale ha anche l’effetto di innescare l'attività idrolasica di GroEl, ccovertendo ATP in ADP, che rilascia. Questo a sua volta causa il rilascio di GroES e l'apetura del poro. Il processo fornisce un “timer” chimico per l’intera operazione.
- Da quando entra nella Gabbia di Anfinsen, la catena polipeptidica ha tempo ca. 20 millisecondi per ripiegarsi correttamente, prima che avvenga l'idrolisi e il coperchio GroES si stacchi, con rilascio della catena a prescindere dal fatto che abbia completato il ripiegamento o no.
- Se questo tempo non è stato sufficiente, viene ripescata in successivi cicli di denaturazione/folding assistito. Se in seguito si ripiega correttamente si avvia a svolgere la sua funzione. Se invece c'è un difetto che impedisce il ripiegamento, sarà eventualmente eliminata dai meccanismi di controllo cellulare.
- Chaperon come GroEL/GroES sono note anche come 'heat shock proteins' (HSP) poichè aiutano a ripiegarsi anche proteine che hanno perso la loro struttura nativa a causa di shock termico. Queste diverse funzioni d sono schematizzate nella Fig. 7.
5) Il rilascio della proteina da un lato di GroEl è intimamente collegata con l'introduzione di un’altra catena proteica nel poro dall’altro lato, ed al legame di ATP alle sue subunità. Il rilascio da un lato è collegato con l'introduzione di una seconda catena dall’altro lato, ed il processo è quindi molto efficiente.
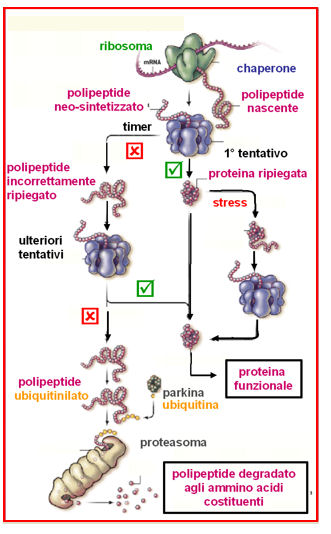
Fig. 7 Processi che coinvolgono le chaperon molecolari