
Macromolecular structure (Inglese)
MACROMOLECULES
This text is divided into five major sections:
- Chemistry of the bonds in biological macromolecules
- Helicity in macromolecules
- Macromolecular folding
- Macromolecular interactions
- Denaturation
- Topology
Introduction
There are three major types of biological macromolecules in biological systems.
- Polysaccharides
- Nucleic acids
- Proteins
Often they are treated separately in different segments of a course. In fact, the principles governing the organization of three-dimensional structure are common to all of them, so we will consider them together.
We will begin with the monomer units.
- monosaccharide -- for carbohydrate
- nucleotide -- for nucleic acids
- amino acid -- for proteins
- We will describe the features of representative monomers, and see how the monomers join to form a polymer.
- We will then look at the monomers in each major type of macromolecule to see what specific structural contributions come from each.
- The three-dimensional structure of each type of macromolecule will then be considered at several levels of organization.
- We will investigate macromolecular interactions and how structural complementarity plays a role in them.
- The concepts for proteins, monosaccharides and nucleotides are just variations on the same theme. So you'll need to learn only one pattern, then apply that pattern to the other systems.
- We will conclude this section of the course with a consideration of denaturation and renaturation -- the forces involved in loss of a macromolecule's native structure (that is, its normal 3-dimensional structure), and how that structure, once lost, can be regained.
1) Biological macromolecules are polar
The main point of the first segment of this material is this:
THE MONOMER UNITS OF BIOLOGICAL MACROMOLECULES HAVE HEADS AND TAILS. When they polymerize in a head-to-tail fashion, the resulting polymers also have heads and tails - they have a directionality.
These macromolecules are polar [polar: having ends with different characteristics] because they are formed by head to tail condensation of polar monomers. Let's look at the three major classes of macromolecules to see how this works, and let's begin with carbohydrates.
A) Monosaccharides condense to yield polysaccharides.
Glucose is a typical monosaccharide. It has two important types of functional group: a carbonyl group (an aldehyde in glucose, some other sugars have a ketone group instead) hydroxyl groups on the other carbons. This is what you need to know about glucose, not its detailed structure.
Glucose exists mostly in ring structures. (5-OH adds across the carbonyl oxygen double bond.) This is a so-called internal hemiacetal. The ring can close in either of two ways, giving rise to anomeric forms, -OH down (the α-form) and -OH up (the β-form).
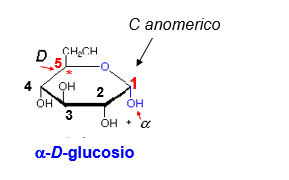
The anomeric carbon differs significantly from the other carbons ( it is the only carbon with TWO oxygens -- ring and hydroxyl -- attached). Free anomeric carbons have the chemical reactivity of carbonyl carbons because they spend part of their time in the open chain form. They can reduce alkaline solutions of cupric salts. Sugars with free anomeric carbons are therefore called 'reducing sugars'. The rest of the carbohydrate consists of ordinary carbons and ordinary -OH groups. A monosaccharide can therefore be thought of as having polarity, with one end consisting of the anomeric carbon, and the other end consisting of the rest of the molecule.
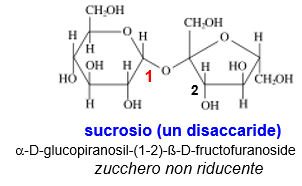
Monosaccharides polymerize by condensation between the anomeric hydroxyl and a hydroxyl of another sugar, forming a 'glycosidic bond'. If two anomeric hydroxyl groups react (head to head condensation) the product has no reducing end (no free anomeric carbon). This is the case with sucrose.
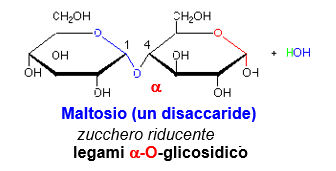
If the anomeric hydroxyl reacts with a non-anomeric hydroxyl of another sugar, the product is polar, with a reducing end (free anomeric carbon)., and a nonreducing end. This is the case with maltose.
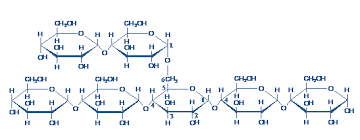
Since most monosaccharides have more than one hydroxyl, branches are possible, and are common. Branches result in a more compact molecule. If the branch ends are the reactive sites, more branches provide more reactive sites per molecule.
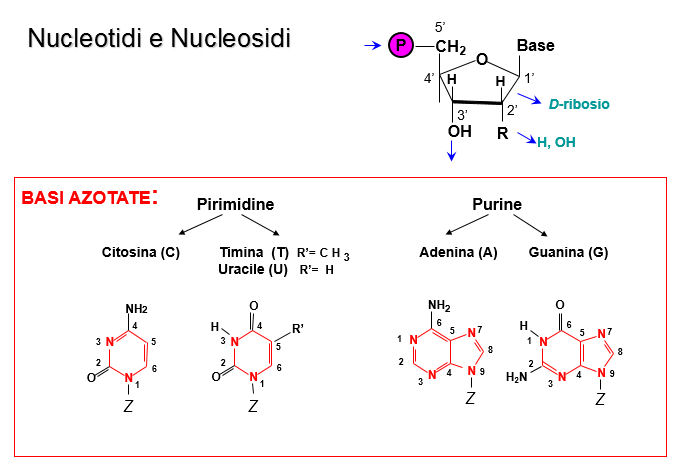
2) Nucleotides condenseto yield nucleic acids.
Nucleotides consist of phosphorylated monosaccharides; ribose (in ribonucleotides), deoxyribose (lacking the 2' -OH, in deoxyribonucleotides) and a base (purine or pyrimidine). The presence or absence of the 2' -OH has structural significance. There are five dominant bases, adenine and gianine (purines), cytosine, thimine and uracyl (pyrimidines)
Uracil and thymine are very similar; differing only by a methyl group. Uracyl is present in ribonucleaotides, thymine in deoxyriboucleotides.
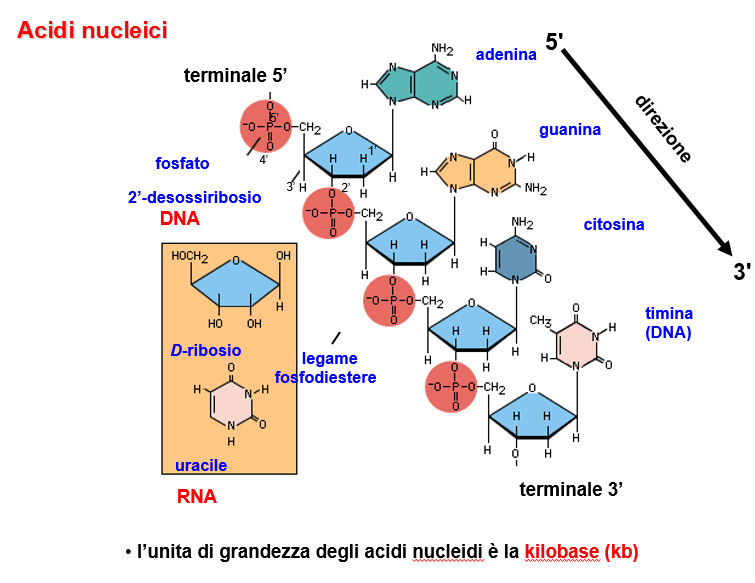
B) Nucleotides condense to form esters between the 5'-phosphate and the 3' -OH of another nucleotide.
A 3'-5' phosphodiester bond is thereby formed. The product is polar as it has a free 5' group (likely with phosphate attached), the 5' terminal, and a free 3' group, the 3' terminal. Bases are abbreviated by their initials: A, C, G and U or T. Sequences are usually written with the5' terminal to the left and the 3' terminal to the right. Phosphate groups are usually not shown unless one wants to draw attention to them.
Branches are possible in RNA but not in DNA, as RNA has a 2' -OH, but branching is very unusual and occurs under very specidfic conditions (e.g. transiently during RNA splicing when the so called "lariat" forms, but not in any finished RNA species).
3) Amino acids condense to form polypeptides or proteins.
Amino acids contain a carboxylic acid (-COOH) group and an amino (-NH2) group attached to a central α-carbon, so they are called α-amino acids. 19 of the nhe naturally occurring amino acids are optically active, as they have four different groups attached to one carbon (glycine is an exception, having two hydrogens) and have the L-configuration.
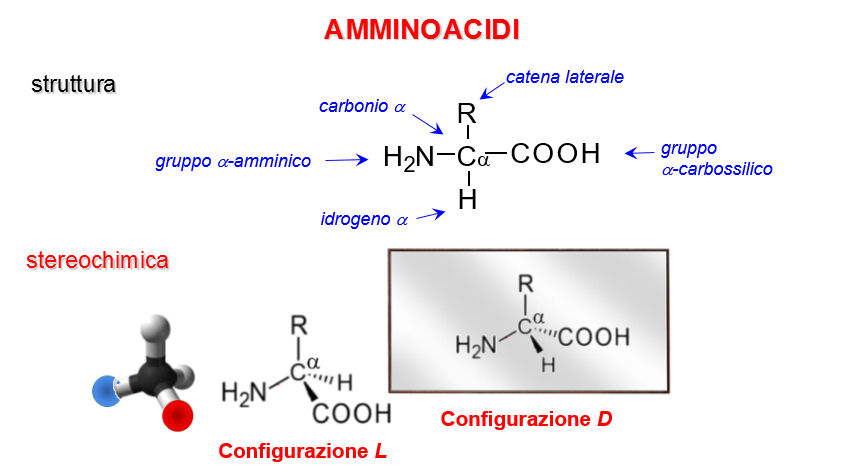
The R-groups of the amino acids (side-chains) provide a basis for classifying amino acids. There are many ways of classifying them,usually it is on the basis of how well or poorly the side-chain interacts with water (hydrophobic or hydrophylic), whether it can form H-bonds, or whether it is charged (see the Amino acids characteristics table)
When aminoacids condense, they form an amide between the carboxyl group of one residue and the amino group of the next. The amide link that is formed is called a peptide bond. The product has ends with different properties; the end with a free amino group is called the a N-terminal, and the end with a free carboxyl groups called the C-terminal.
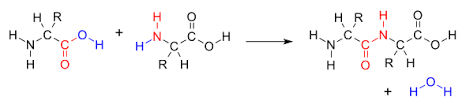
peptide bond
The aminoacid residues are referred to by three letter abbreviations, that are are self-evident, such as Gly for glycine or Asp for aspartate, etc. The one-letter abbreviation system is also heavily used to save space in databases and in articles. You must knnow these abbreviations and you can find them in the Amino acids characteristics table.
By convention, sequences are written with the N-terminal to the left and the C-terminal to the right. Although the seide-chains of some amino acids contain amino and carboxyl groups, branched polypeptides or proteins do not occur. The sequence of monomer units in a macromolecule is called the PRIMARY STRUCTURE of that macromolecule. Each specific macromolecule has a unique primary structure. When proteins fold, sections of sequence adopt a regular conformation, and this is known as SECONDARY STRUCTURE. These elements of 2y structure then associate to from the TERTIARY STRUCTURE - the native, functional structure of the protein in which all atoms have a place in 3-D molecular space. Once they have adopted their 3y structure, protein subunits can then aggregate in a precise manner to form complexes with QUATERNARY STRUCTURE.
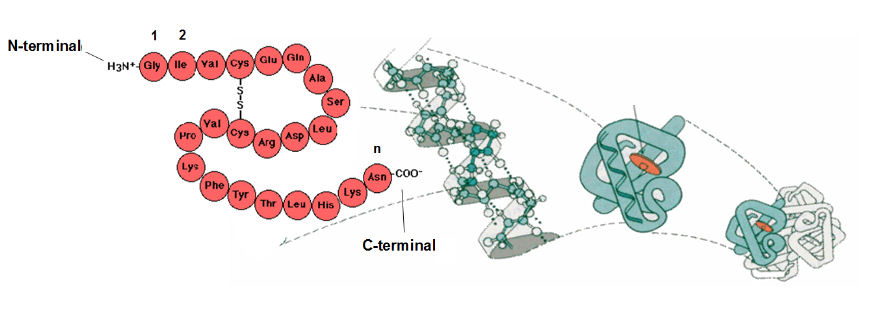
Structural hierarchy: 1y 2y 3y 4y
Biopolymers consisting of regularly repeating units tend to form helices.
The fundamental reason for this is that the bond angles (torsional angles) of the constituent atoms are almost never 180 degrees (completely extended trans conformation), so linear molecules are not very likely. Angles less than 180° result in a more or less gentle curving of the molecule (it becomes spiral).
What exactly is a helix? One definition is that a helical structure consists of repeating units that lie on the wall of a cylinder such that the structure is superimposable upon itself if moved along the cylinder axis. For this reason, a helix looks like a spiral, or has a screw-like appearance. A chain with a zig-zag appearance is a degenerate helix.
Helices can be right-handed or left handed, depending on if they have a clockwise or anticlockwise movement of the chain. Helical organization is an example of secondary structure. These helical conformations of macromolecules persist in solution only if they are stabilized by internal H-bonds.
Stable helices in biomoleculesare usually maintained by intramolecular hydrogen bonds.
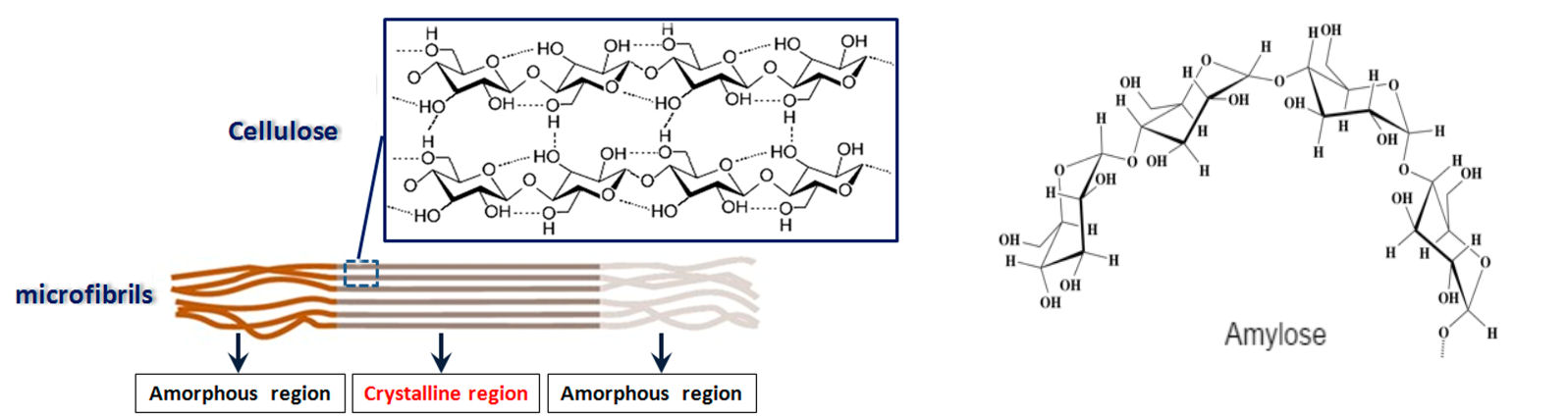
Helices in carbohydrates. Polysacchardes with long sequences of α(1 -> 4) glycosidic links have a weak tendency to form helices. Starch (amylose) exemplifies this, so that the starch helix is not very stable and it adopts a random coil conformation in solution. β(1 ->4) sequences favor linear structures. Cellulose exemplifies this and can be considered as a degenerate helix consisting of zig-zagging glucose units in alternating orientation, stabilized by intrachain hydrogen bonds. Cellulose chains lying side by side form sheets of linear molecules stabilized by interchain hydrogen bonds.
Helices in proteins. Properties of the peptide bond have a marked effect on the structures of proteins.
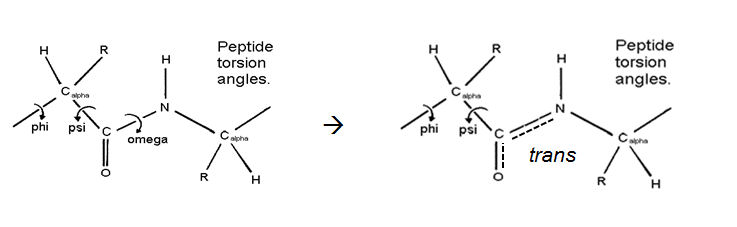
- The first major property of the peptide bond is that it has a partial double-bond character, conferred by delocalization of electrons towards the electronegative carbonyl oxygen, which draws the unshared electron pair from the amide hydrogen. As a result of its double bond character, the peptide bond is planar, not free to rotate and prefers the the trans configuration. These characteristics restricts the conformational space accessible to the polypeptide chain,
- The second major property of the peptide bond is that N and O atoms can form H-bonds. These will try to form the highest possible number of H-bonds
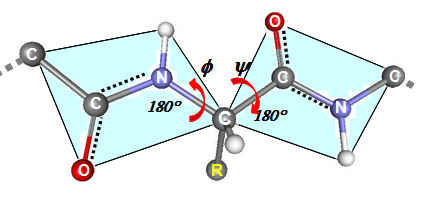
- The third major property of the peptide bond is that although it is rigid and planar. the bonds flanking it in the polypeptide have rotational freedom. The torsional angle φ (Phi) about the -N-αC- bond, and ψ (Psi) angle about the -αC-CO- bond allow the polypeptide chain to adopt different conformations. In particular, if φ has a value ~ -60° and ψ a value ~ -45-50° there is little steric hindrance among the atoms forming the polypeptide main chain. Also if the angles are 120-180° (extended conformation - usually 120-160°) there is little steric hindrance among the atoms forming the polypeptide main chain.
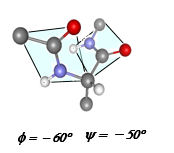
The α-helix is a major structural component of proteins.
Stabilizing factors include:
- H-bonds are formed between peptide all C=O and N-H groups along the backbone (between C=O in position i and H-N in position i+4)
- these H-bonds are all between different parts of the same helical segment of the chain (i.e. intra-segment H-bonds, the conformation is self-stabilizing)
- as many H-bonds form in this way, they strongly stabilide the conformation
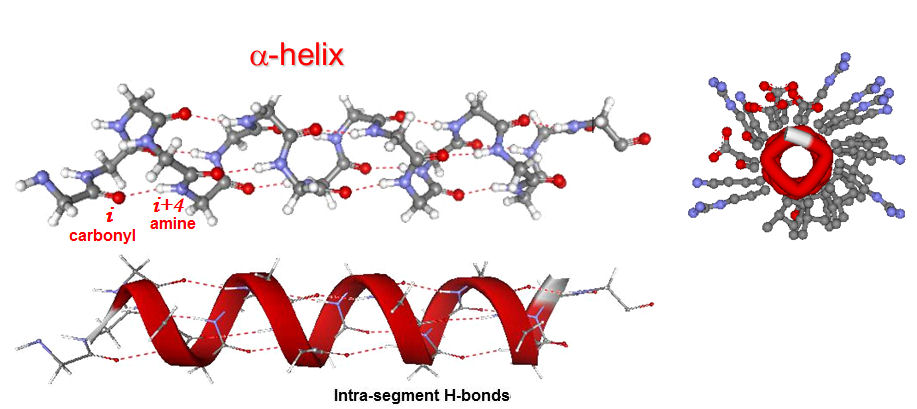
The α-helical conformation frequently occurs as the component of globular proteins, but also as a component of some fibrous proteins, like α-keratin, which has a high tensile strength (found in hair, feathers and horn). Keratin is formed by helical polypeptides wound around each other to form a rope-like fiber, linked by intramolecular H-bonds and disulfide bridges.

Globular protein (myoglobin) Fibrous protien (α-keratin)
The β-pleated sheet is a second major structural component of proteins.
β-sheets consist of extended sections of peptide chains with a zig-zag arrangement -- degenerate helices -- known as β-strands. These strands lie side by side and are hydrogen bonded to one another (unlike in α-helices where H-bonds are intra-segment, in β-sheets H-bonds are inter-segment). The polypeptide chains of a β-sheet can be arranged in two ways: parallel (running in the same direction) or antiparallel (running in opposite directions). An edge-on view shows the pleats.
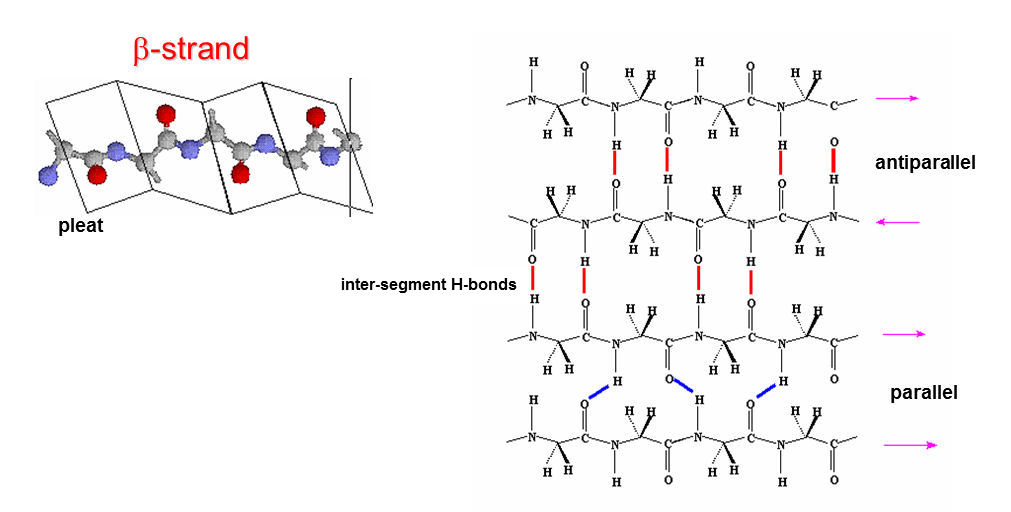
Stabilizing factors for the β-sheet resemble those for the α-helix.
- β-pleated sheets are found as a component of 80% of all globular proteins. They are often represented as ribbons, sometimes with an arrow head showing the direction of the chain (N- to C-terminal). They are usually present as distorted sheets. Sometimes the sheet bends completely to form a 'β-barrel'. Some transmembrane proteins (e.g. porins) are very large β-barrels. β-pleated sheets are also found in some fibrous proteins, such as in silk fibroin. In this case the sheets are relatively flat and stacked one over the other.

- Globular β-sheet protein Globular β-barrel protein Transmembrane β-barrel
- - All possible H-bonds form between peptide C=O and N-H groups in the backbone , however in this case the H-bonds are all inter-segment, unlike those of the alpha-helix, where they are intra-segment.
- - All peptide bonds are trans and planar. Side-chains are oriented above and below the plane of the sheet in an alternating manner. Small side-chains are less destabilizing than large side-cahins due to steric hindrance
- Fibroin, a fibrous β-sheet protein
Turns and Loops
Other elements of secondary structure are necessary to join sections with regular conformations.
- these conformational elements allow the chain to change direction between elements of regular secondary structure (α-helices and β-strands).
- In some cases they are 'tight turns" involving only 2 or 3 aminoacid residues
- In other cases they are 'wide loops' involving 6 or more residues
Turns have well defined conformations and can fall into the β (3 residue) or γ types. H-bonding occurs within the turn itself. There are three different types of β-turns, where the the peptide i carboxyl oxygen forms a H-bond with the i + 3 amide nitrogen. In γ-turns the peptide i carboxyl oxygen forms a H-bond with the i + 2 amide nitrogen. Gly and Pro residues are often prresent in turns, due to their particular steric characteristics.
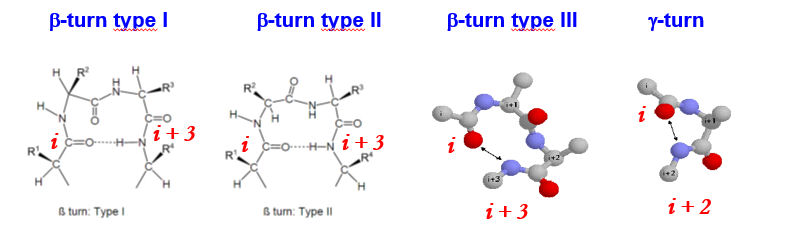
Wheter two elements of 2y structure are connected by a tight turn or a wide loop depends on steric factors. β-strands can come quite close to each other side-by-side, as the side-chains point up and down. This means that two antiparallel -strands can be connected by a tight turn. This gives rise to the β-hairpin motif.
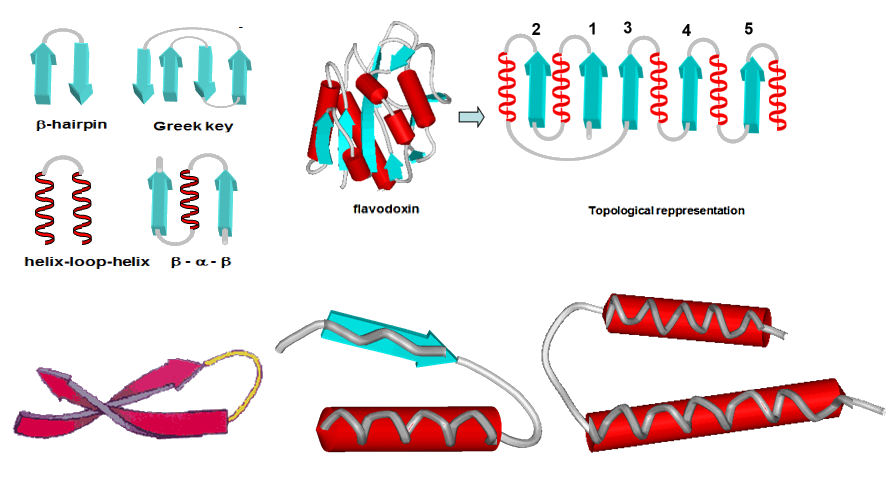
α-helices are bulky, as the side-chains are pointing outwards in all directions (see above), so that they must be connected by wide loops (helix-loop-helix motif). For the same reason, β-strands are connected to α-helices by wide loops ( αβ motif).
2y structure elements are connected to each other in a limited number of different ways. These patterns can occur in proteins with quite different structures and functons and are known as TOPOLOGICAL elements or SUPERSECONDARY structure
Topological elements and Supersecondary structure.
These are patterns of 2y structure elements connected by turns or loops that and are repeated in different proteins. Proteins can have a similar topology even though the actual 3y structure is quite different. This is like two maps being topologically similar, even though they refer to completely different landscapes. A few common topological elements are the β-hairpin, helix-loop-helix, the β-α-β motif, the four-helix bundle, and the Greek key. This takes its name from the similarity to the pattern seen on greek temples.
TERTIARY STRUCTURE
CONCEPT:
- NUCLEIC ACIDS AND PROTEINS ARE LARGE MOLECULES WITH COMPLICATED THREE-DIMENSIONAL STRUCTURES.
- THESE STRUCTURES ARE FORMED FROM SIMPLER ELEMENTS of 2y STRUCTURE
- THE 3y STRUCTURE DESCRIBES THE EXACT POSITION OF ALL ATOMS IN THE STRUCTURE
- SOME GENERAL PATTERNS DESCRIBE THE OVERALL ORGANIZATION FOR MOST MACROMOLECULES.
Tertiary structure in Proteins
The formation of compact, globular structures is governed by the constituent amino acid residues. Folding of a polypeptide chain is strongly influenced by the hydrophobicity/hydrophilicity amino acid side-chains (see AA characteristics table).
- Hydrophobic side-chains, as in leucine and phenylalanine, normally orient inwardly, away from water or polar solutes.
- Polar or ionized side-chains, as in glutamine or arginine, orient outwardly to contact the aqueous environment.
- Some amino acids, such as glycine, can be accommodated by aqueous or nonaqueous environments.
The rules of hydrophobicity and the tendency for secondary structure formation determine how the chain spontaneously folds into its final structure.
- Forces stabilizing protein tertiary structure.
- Hydrogen bonding, as part of any secondary structure, as well as other hydrogen bonds.
- Ionic interactions -- attraction between unlike electric charges of ionc side-chains.
- Disulfide bridges between cysteinyl residues. The side-chains of cysteine is -CH2-SH. -SH (sulfhydryl) groups can oxidize spontaneously to form disulfides (-S-S-). R-CH2-SH + R'-CH2-SH + O2 = R-CH2-S-S-CH2-R' + H2O2 (Under reducing conditions a disulfide bridge can be cleaved to regenerate the -SH groups.)
The disulfide bridge is a covalent bond. It strongly links regions of the polypeptide chain that could be distant in the primary sequence. It forms during tertiary folding or after it has occurred, so it stabilizes but does not determine tertiary structure.
Globular proteins are typically organized into one or more compact patterns.
Proteins often have autonomously folding regions kinown as domains. It is also found that certain structural themes occur in different proteins, and often, but not always, in proteins that have similar biological functions. This phenomenon of common structures is consistent with the notion that many proteins are genetically related ( they derive from a common ancestor). The amino acid sequences of these proteins sometimes show a significant homology, so it could be predicted that their 3-D structures are similar. On the other hand, proteins with very similar 3-D structures may have no sequence similarity.
There are a few structural themes that occur quite often in proteins. These are referred to as topological elements, or elements of SUPER-SECONDARY structure. a few examples are:
The four-helix bundle (see above)
This structural element is a common pattern found in globular proteins. Helices lying side by side can interact favorably if the properties of the contact points are complementary. Hydrophobic amino acids (like leucine) at the contact points and oppositely charged amino acids along the edges will favor interaction. If the helix axes are inclined slightly (18 degrees), the side-chains will interdigitate perfectly along 6 turns of the helix. Sets of four helices yield stable structures with symmetrical, equivalent interactions. Interestingly, as four-helix bundles diverge at one end, they can provide a cavity in which ions may bind.

Four-helix bundle
All-β domains
These also occur in many globular proteins. β-pleated sheets can fold back on themselves to form barrel-like (see above) or sandwich-like structures. The immunoglobulins are a good example of a protein with sandwich-like all-β domains.
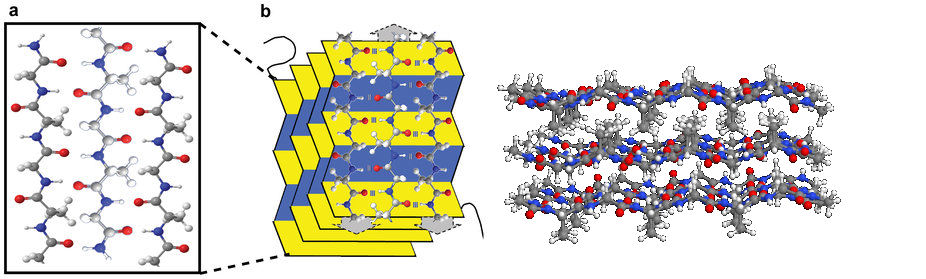
Unfortunately, sometimes proteins can change from their native conformation to an all-β structure, and this catalyses the same transition in other protein molecules. The sheets then start to stack (associate) and form larger and larger oligomeric complexes that can be very damaging to cells (e.g. amyloid fibers).
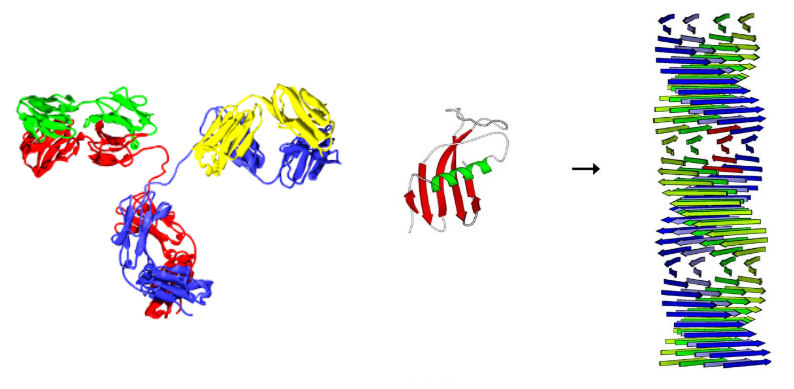
Immunoglobulin Transition of a β-sheet to an amyloid-like fiber
α/β - domains.
These are found in different proteins independently of their function. These domains are organised in alternating β-α-β (see topological elements above) structures that can give rise to β-barrel type proteins (see above), where the presence of the long helix allows the β-sheets to arange in a parralel manner. Alternatively, the helices may be on both sides of a single distorted sheet, forming a sandwich-like structure.
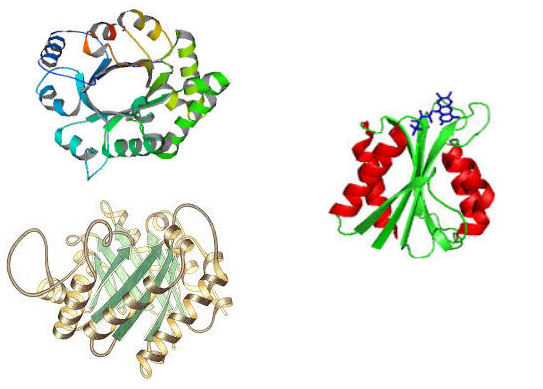
- α/β-barrel (triose phosphate isomerase) α/β sandwich (flavodoxin)
Macromolecular Interactions
CONCEPT:
- MACROMOLECULES INTERACT WITH EACH OTHER AND WITH SMALL MOLECULES.
- THESE INTERACTIONS REQUIRE COMPLEMENTARITY BETWEEN THE INTERACTING SPECIES.
- OFTEN, THIS REQUIRES AN EXACT FIT OF SIZE, SHAPE AND CHEMICAL CHARACTERISTICS .
Macromolecular interactions involving proteins - QUATERNARY STRUCTURE.
Quaternary structure refers to proteins formed by association of independent polypeptide subunits. Individual globular polypeptide subunits associate to form biologically active oligomers (Oligo = several; mer = body)
The association is specific:- if a limited number of subunits is involved, it is known as a 'closed' 4y structure
- 2 (dimer) and 4 (tetramer) subunits are most common, but trimers, pentamers, etc. also occur
- the subunits may be identical or they may be different. (homo- or hetero-oligomers)
- subunit interaction are noncovalent between complementary regions of subunit surface
- hydrophobic regions favour interaction
- H-bonding favours precise interaction (it is directional)
- electrostatic (ionic) attraction also favour interaction (they help docking of subunits as they are relatively long-range).
If covalent links exist (such as disulfide bridges) then the structure is technically not considered quaternary, as the subunits are connected by a coalent bond. In proteins with quaternary structure the deaggregated subunits alone are generally biologically inactive. Quaternary structure in proteins is the most intricate degree of organization considered to be a single molecule. Higher levels of organization are known as multimolecular or supramolecular complexes.
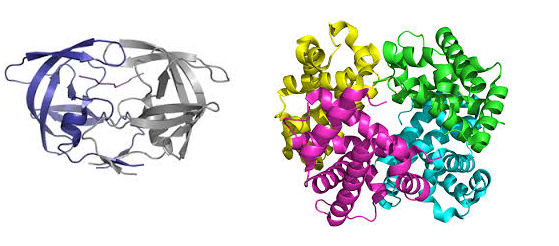
HIV aspartic protease (homodimer) Haemoglobin (heterotetramer)
Incorporation of nonprotein components into proteins
If moieties that are not aminoacids are incorporated into proteins, they are called conjugated proteins. We can therefore establish two categories of proteins:
- Simple proteins consist only of polypeptide subunits .
- Conjugated proteins also contain a nonprotein moiety which frequently plays a role in biological function. It may be required directly for the function or it may be required for the correct structure of the conjugate protein.
- - Heme
- Lipids
- Carbohydrates (monosaccharides, oligosaccharides or polysaccharides)
- Metal ions (most often Zn, Fe, Cu and Mg but also Mn, V, Mo etc.)
- Phosphate groups
Nomenclature:
- apoprotein = the protein without its nonprotein component
- holoprotein (conjugated protein) = the apoprotein + prosthetic group.
- prosthetic group = the nonprotein entity
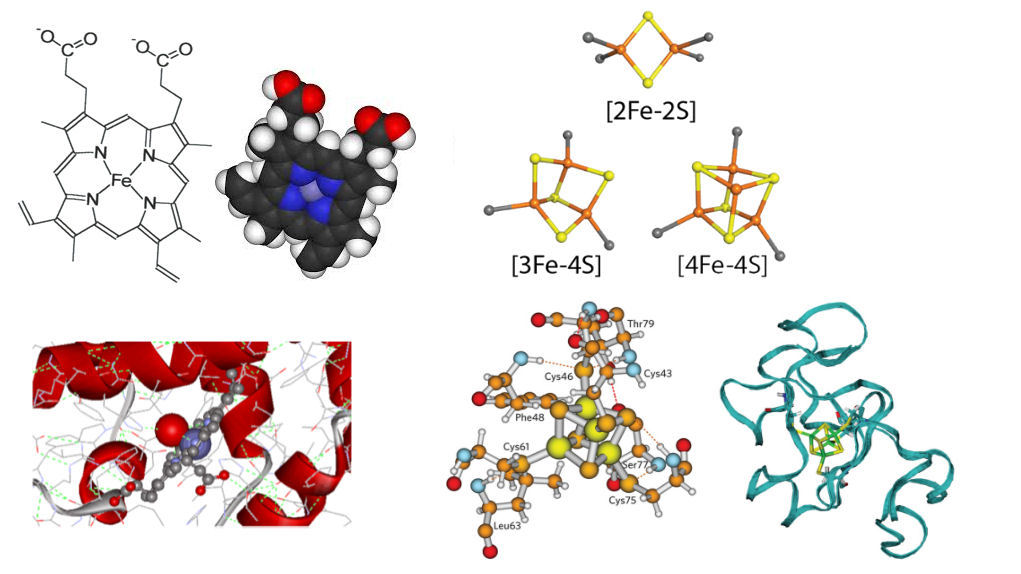
Metalloproteins
Metals found as prosthetic groups of proteins include Mg, Ca, V, Cr, Mn, Fe, Co, Cu, Zn and Mo. (Also W in some archaeobacteria). These metals can form coordination complexes, as they accept electron pairs from atoms with unshared electron pairs (the electron pairs fill vacant orbitals of the metal ion, such as sp3d2 orbitals). Some of these metals can easily undergo oxidation-reduction [e.g Fe(II) = Fe(III) + e]. All these metals are relatively small; no heavy metals (e.g., Pb, Hg) are included.
- Structural - by complexing several side-chains in the protein simultaneously, they stabilizing the three-dimensional structure of the protein (e.g. Ca in calmodulin, Zn in aspartate transcarbamylase)
- Positioning - by binding the protein to another molecule (e.g., a substrate) they can help in the catalytic process (e.g. Zn in Carboxypeptidase).
- Functional - e.g. they can be part of the catalytic process of the enzyme, such as activation of a substrate. When a metal accepts an electron pair form a bound substrate, the resulting electron deficiency may make the substrate more reactive.
- Chemical - Metals can participate in oxidation-reduction reactions. Sometimes bound metals participate directly in biological oxidation-reduction reactions by accepting or donating an electron (changing oxidation state).
aspartate transcarbamylase calmodulin myoglobin
Sometimes other organic or inorganic compounds share metals with proteins.
- Sulfide ions participate in formation of the iron-sulfur centers of redoxins (FenSm centers).
- Heme -- here the iron is part of a large organic complex. It is bound by coordination links to the organic moiety. Binding to the protein partly through one of the remaining coordination links partly through the organic moiety (e.g. myoglobin).
Lipoproteins
Lipoproteins are transport systems for lipid molecules. These protein associate with lipids through non-covalent hydrophobic interactions involving the protein' aminoacid side-chains and lipid acyl chains. Lipoproteins are 'pseudomicellar' structures. Micelles are orderly arrays of lipid molecules having polar heads and hydrophobic tails. In water, the polar heads orient outward, and the polar tails cluster in the center of the micelle. Lipoproteins resemble micelles in some respects.
The structure of lipoproteins typically includes the following features.
- The outer surface is formed by polar lipid head groups and cholesterol, with protein inserted into it.
- The interior is a region of randomly oriented hydrophobic lipid acyl chains and neutral lipids (e.g. triacylglycrols, cholesterol esters).
- Lipoproteins are usually quite large structures.
- The role of the polar lipid and protein on the surface is to solubilize the neutral lipid interior.
- The protein interacts with the lipid of lipoproteins through amphipathic helices. α-helical regions of apolipoproteins have polar amino acids on one surface, and nonpolar ones on the opposite surface. The helix lies on the surface of the structure, with the polar groups oriented outward toward the water, and the nonpolar groups buried in the lipid. (Recall the four-helix bundle domains of proteins, in which contacts between helices involved hydrophobic residues at the contact points.).
- The protein on its own is known as apolipoprotein.
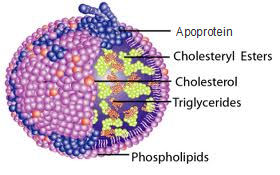
lipoprotein particle
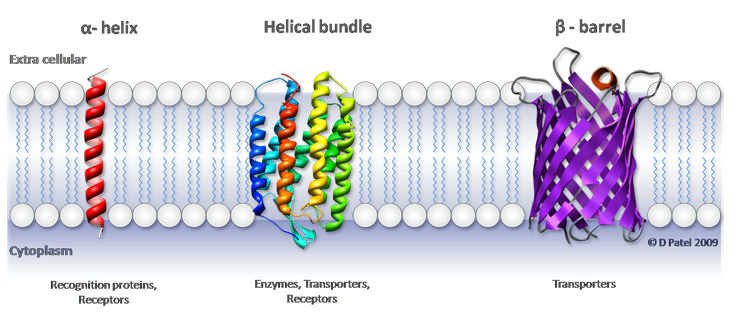
Membrane proteins
These are in some ways similar to apolipoproteins as that they have non-polar amino acids in strategic locations to permit interaction with the membrane lipid. Proteins of the membrane surface may be structured like the apoproteins of lipoproteins, with amphipathic helices.
Some membrane proteins transverse the entire membrane. The region of the protein that is completely immersed in membrane should consist entirely of hydrophobic amino acids (transmembrane or TM domain). A common structural motif to accomplish this is an α-helix.
Some TM proteins have a transmembrane region consisting of a single helix of at least 22 hydrophobic aminoacids. This makes the α-helix long enough to span a membrane (~2nm).
Other TM proteins have arrays of membrane-spanning helices (typically 7 or 12), and in this case the helices can be shorter.
Another common motif of TM proteins is the β-barrel.
Glycoproteins
These are proteins with carbohydrate prosthetic groups.
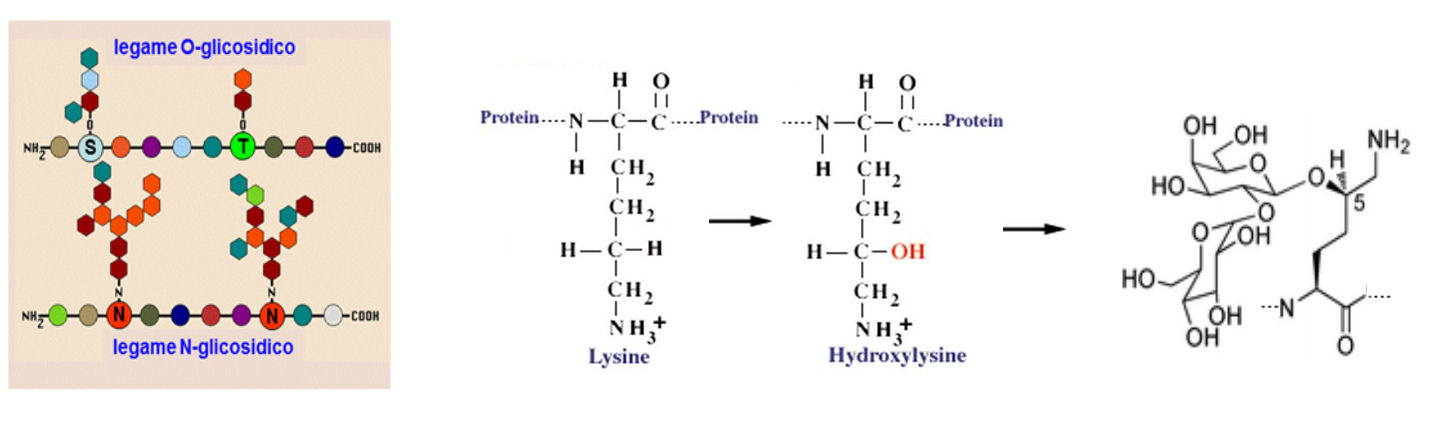
Typical structure -- 1 to 30 carbohydrate residues can form chains that covalently link to the apoprotein in one of three major ways. The oligosaccaride may be straight or branched.
- N-linked to Asn (Type I) N-acetylglucosamine (a sugar with an acetylated amino group in place of a hydroxyl group) at the reducing end of a carbohydrate chain is linked to the amide nitrogen of asparagine residue. The asparagine residue must be in the sequence, Asn X Thr (or Ser), where X is any amino acid residue. This specific sequence is called a sequon. No other asparagine will bind oligosaccharide.
- O-linked Ser/Thr (Type II): Here the reducing end of a carbohydrate chain (usually N-acetylglucosamine residue) is linked to the hydroxyl of a Ser or Thr residue.
- O-linked to Hyl (Type III) in collagen: In this case The reducing end of a carbohydrate chain (usually N-acetylgalactosamine) is linked to the hydroxyl of a hydroxylysine residue in collagen. Hydroxylysine (Hyl) is produced enzymatically from lysine in collagen after the collagen has been synthesized.
Glycoproteins have two major types of functions.
- Recognition: carbohydrate prosthetic groups serve as antigenic sites (e.g., blood group substances are carbohydrate prosthetic groups), intracellular sorting signals (mannose 6-phosphate bound to a newly synthesized protein sends it to the lysosomes), etc.
- Structural
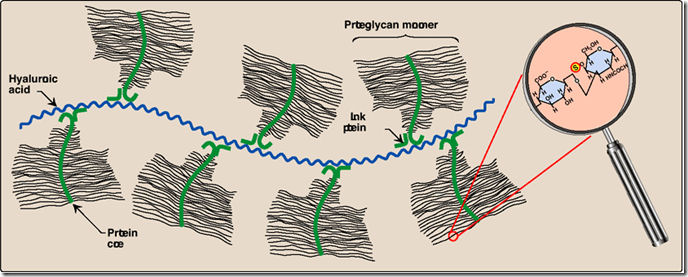
a) Proteoglycans of cartilage, for example, have polysaccharide central core called hyaluronic acid. Many glycoprotein branches are attached to the hyaluronic acid noncovalently. Each branch is a glycoprotein (core protein) with many carbohydrate chains (chondroitin sulfate -- alternating galactosamine and galactose -- and keratan sulfate -- alternating glucosamine and galactose) attached covalently (xylose beta-> O-ser). The attachment of the core protein to the hyaluronic acid is mediated by a protein called the link protein.
b) extracellular proteins are often glycosilated as it stabilizes the structure and makes them more soluble.
Proteoglycan structure
Interactions between proteins and nucleic acids.
This type of interaction is based on structural complementarity. There are two general types of interactions to consider:
- Protien/nucleic acid complexes have both polypeptide and nucleic acid subunits. A good example are the ribosomes or other type of riboproteins (complexes of polypeptides and RNA strands).
- DNA-binding proteins. Proteins that bind to double stranded (ds) DNA and carry out several diffferent types of processes, such as strand cleavage (nucleases), winding or unwinding the double helix (isomerases), or controlling gene expression (regulatory proteins).
Ribosome are complex molecular machines found in all cells that carry out protein synthesis (translation). They join amino acids together in a specific order using information carried by the messenger RNA (mRNA) molecules.
Structurally, ribosomes have two major components. The small ribosomal subunit (30S) reads the mRNA, and the large subunit (50S) catalyses the synthesis of the polypeptide chain. Each subunit is composed of one or more ribosomal RNA (rRNA) strands and a several polypeptide strands. When the 30S and 50S subcomplexes come together they form the fully functional 70S ribosomes. 30S, 50S and 70S refer to the sedimentation speed during ultracentrifugation (an experimental technique used to study macromolecules) and is related to the size of the subcomplex.
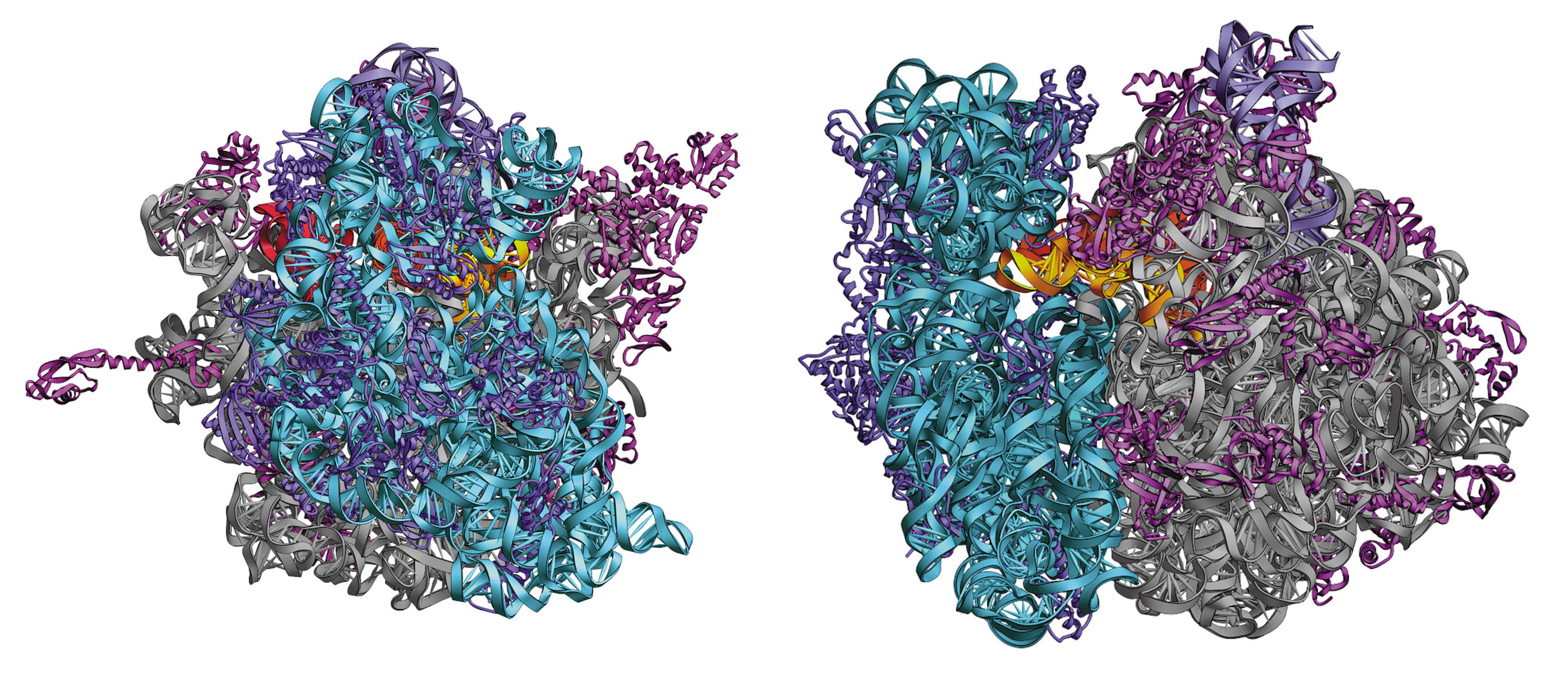
Complete 70S ribosome, the 30S subuit is in blue
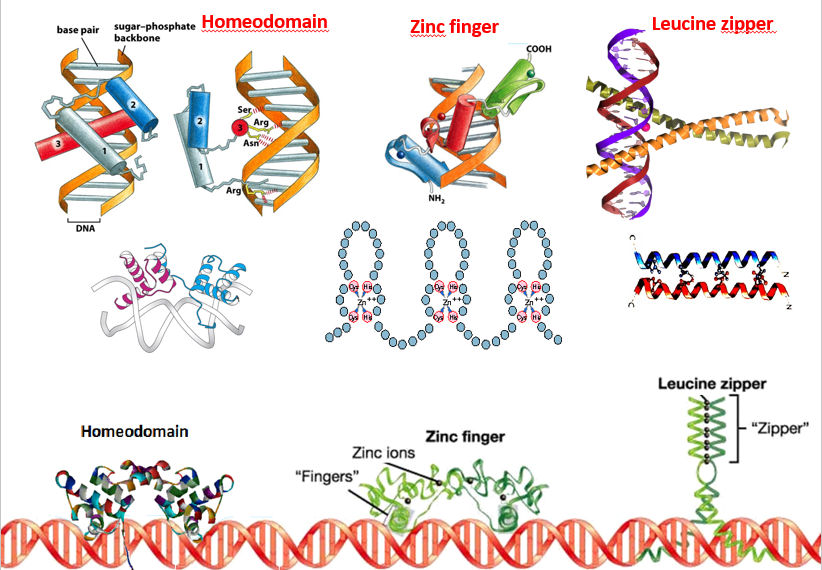
DNA-binding proteins
There are three characteristic motifs present in DNA-binding proteins
The zinc finger motif: A small Zn-stabilized structural domain found in proteins that interact with nucleic acids. The zinc finger is a loop of about 25 aminocid residues stabilized by a Zn atom.
- Zn forms coordination bonds to His and/or Cys , which maintains the structure of the domain.
- Unlike a -S-S- bridge, the Zn complex will not be broken by reducing conditions within the cell
- Unlike Cu or Fe, Zn does not participate in oxidation-reduction reactions that could generate free radicals which might damage nucleic acids.
Other aminoacid residues in the loop are involved in binding to specific nucleotides of the nucleic acid or helping to maintain the folded structure of the domain. Zinc fingers in proteins occur in tandem arrays. They are joined to nearby zinc fingers by short linking regions of peptide. They are spaced to fit into the major groove of DNA, with the bases of the alpha-helices down in the grooves, and the beta-loops touching the double helix.
The leucine zipper: A pair of amphipathic alpha-helices joining two subunits of a dimeric protein that binds to DNA. Some sites in DNA important to biological control have twofold symmetry: the base sequence is the same in both directions.
- Example:
- 5' ...TGACTCA... 3'
- 3' ...ACTGAGT... 5'
A protein designed to bind at such a site should also be symmetric. A class of DNA-binding proteins that forms symmetric dimers using amphipathic α-helices that have regularly spaced leucine residues along one edge. They have some similarity to 4-helix bundle, but with just two helices. Leucine residues face each other along the two interacting helices like the teeth in a zipper. The symmetric dimer then binds to the symmetric region of the DNA through special binding domains.
The helix-turn-helix motif: Short adjacent α-helices that cross one another. One α-helix fits into the major groove of DNA, and interacts with specific bases; this is called the recognition helix. A loop or turn links the recognition helix to a second helix which is connected to a third helix, both of which lie across the major groove of DNA and bind nonspecifically. The helix-turn-helix motif is another common motif in proteins.
A dimeric protein can have a helix-turn-helix motif in each subunit, and if the monomer units are identical it can recognize and bind to symmetric DNA structures. The type of DNA-binding protein that use this motif are known as homeodomain proteins
Denaturation (see also page on Folding)
CONCEPT:
- THE DISRUPTION (DENATURATION) OF A MACROMOLECULE'S THREE-DIMENSIONAL STRUCTURE REQUIRES DISRUPTION OF THE FORCES RESPONSIBLE FOR ITS STABILITY.
- THESE FORCES ARE THOSE THAT STABILISE THE MACROMOLECULE (HYDROPHOBIC INTERACTIONS, H-BONDS, VDW INTERACTIONS AND ELECTROSTATIC INTERACTIONS)
- WHEN A FEW OF THESE START TO BREAK, IT FACILITATES THE RUPTURE OF MORE NON-COVALENT BONDS, SO THE PROCESS IS COOPERATIVE.
- THE ABILITY OF PHYSICAL (TEMPERATURE) OR CHEMICAL (IONS, CHAOTROPIC) AGENTS TO HELP ACCOMPLISH THIS DISRUPTION CAN BE PREDICTED ON THE BASIS OF WHAT IS KNOWN ABOUT MACROMOLECULAR STABILIZING FORCES.
- DENATURED MACROMOLECULES WILL USUALLY RENATURE SPONTANEOUSLY (UNDER SUITABLE CONDITIONS), SHOWING THAT THE MACROMOLECULE ITSELF CONTAINS THE INFORMATION NEEDED TO ESTABLISH ITS OWN THREE-DIMENSIONAL STRUCTURE.
Denaturation is the loss of a protein's or DNA's three dimensional structure (its NATIVE structure). Denaturation is physiological event -- biological structures should not to be too stable. Examples of NORMAL macromoleclar denaturation are:
- Double stranded DNA must come apart to replicate and for RNA synthesis (transcription).
- Proteins must be degraded under certain circumstances (e.g. because they become senescent, or to terminate their biological action, as in enzymes).
- To release amino acids (e.g.,as substrates for gluconeogenesis in starvation).
Loss of native structure must involve disruption of factors responsible for its stabilization. These factors are:
- H-bonding
- Hydrophobic interaction
- Electrostatic interaction
- Disulfide bridging (in proteins)
Note that no break in the polymer chain (disruption of primary structure) is involved in denaturation. Molecules that disrupt these stabilizing factors are known as Denaturing agents and mainly disrupt H-bonding.
Factors that cause denaturation
- Heat --> kinetic agitation (vibration, etc.) -- will denature proteins or nucleic acids. Heat denaturation of DNA is called melting because the transition from native to denatured state occurs over a narrow temperature range. As the purine and pyrimidine bases become unstacked during denaturation they absorb light of 260 nanometers wavelength more strongly. The abnormally low absorption in the stacked state is called the hypochromic effect. This means that DNA denaturation can be followed spectrofotometrically.
- Chaotropic agents - Urea and guanidinium chloride -- Thse work by competition for H-bonds, as they contain functional groups that can accept or donate hydrogen atoms . At high concentration (8 to10 M for urea, and 6 to 8 M for guanidinium chloride) they compete favorably for the hydrogen bonds of the native structure. For example, H-bonds in an α-helix or β-sheet will be replaced by H-bonds to urea and these secondary structure elements will be disrupted.
- Detergents/solvents - these agents disrupt hydrophobic interaction [e.g. detergents like SDS - sodium dodecyl (lauryl) sulfate, or organic solvents such as acetone or ethanol -- dissolve nonpolar groups).
- Cold -- increases solubility of nonpolar groups in water. When a hydrophobic group contacts water, the water dipoles must solvate it by forming an orderly array around it. The array is called an "iceberg," because it is an ordered water structure, but not true ice. The ordering of water in an "iceberg" decreases the randomness (entropy) of the system, and is energetically unfavorable. If hydrophobic groups cluster together, contact with water is minimized, and less water must become ordered. This is the driving force behind hydrophobic interaction. (The clustering together of hydrophobic groups is also entropically unfavorable, but not as much so as "iceberg" formation). At low temperatures, solvation of hydrophobic groups by water dipoles is more favorable. The water molecules have less thermal energy. They can "sit still" to form a solvation "iceberg" more easily. The significance of cold denaturation is that cold is not a stabilizing factor for all proteins. Cold denaturation is important in proteins that are highly dependent on hydrophobic interaction to maintain their native structure.
- ionic strength or pH - variations in ionic strength or pH extremes can both disrupt electrostatic interactions, Most macromolecules are electrically charged,, as the ionizable groups of the macromolecule contribute to its net charge (sum of positive and negative charges). Electric charges of the same sign repel one another. If the net charge in the interior of a macromolecule is zero or near zero, electrostatic repulsion will be minimized. A compact three-dimensional structure will be favored, because repulsion between parts of the same molecule will be minimal. pH extremes result in large net charges on most macromolecules, if they contain basic or acidic groups. At low pH all the acidic groups will be nutral and basic groups will be positivley charged, so the net charge on the protein will be positive. At high pH all the acidic groups will be negatively charged and the basic groups neutral, so the net charge on the protein will be negative. Intramolecular electrostatic repulsion from a large net charge will favor an extended conformation rather than a compact one.
- Agents that disrupt disulfide bridges -- These agents can interact with free sulfhydryl groups, and will tend to reduce (and thereby cleave) disulfide bridges, so that they can form links to the free sulfhydryls, or between themselves. Some proteins are stabilized by numerous disulfide bridges; cleaving them renders these proteins more susceptible to denaturation by other forces.
Example:
2 HO-CH2-CH2-SH + R1-S-S-R2 = R1-SH + HS-R2 + HO-CH2-CH2-S-S-CH 2-CH2-OH
Renaturation is the regeneration of the native structure of a protein or nucleic acid. Renaturation requires removal of the denaturing conditions and restoration of conditions favorable to the native structure. This includes:
- Solubilization of the substance if it is not already in solution.
- Adjustment of the temperature.
- Removal of denaturing agents by dialysis or similar means (e.g. rapid mixing to reduce the concentration).
- In proteins, re-formation of any disulfide bridges.
Methods for follwing denaturation or folding
- The formation of base-pairs in DNA reduces the absorption of its purine and pyrimidine bases at 260 nm wavelength. This means that denaturation causes an increase in absorption and folding into a double strand cuases a decrease in absorption. These processes can therefore be followed using a spectrophotometer.
- In proteins, tryptophane residues can absorb and then emit light - they are flourescent. This flourescence is very sensitive to the aquoeus environment. If the Trp residues are exposed to water, the flourescence is low. When the protein folds, Trp residues tend to transfer into the organic core of the protein, as they are hydrophobbic. Here they are protected from quenching by water molecules, and the fluorescence becomes stronger. This means that the folding/renaturation process can be followed using a spectrofluorimeter
- In proteins, α-helices and β-sheets have characteristic circular dichroism spectra (see page on CD). This means that the formation of these secondary structural elements can be followed during folding using a dichrometer.
Usually considerable skill and art are required to accomplish renaturation. The fact that renaturation is feasible demonstrates that the information necessary for forming the correct three-dimensional structure of a protein or nucleic acid is encoded in its primary structure, the sequence of monomer units. But...
Folding may be slow and the chain can interact with other chains to aggregate. Guidance may be needed for it to occur correctly and rapidly. Molecular chaperones are intracellular proteins which guide the folding of proteins, preventing incorrect molecular interactions (see page on Folding). They do NOT appear as components of the final structures. Chaperones are widespread, and chaperone defects are believed to be the etiology of some diseases.